

Introduction
Peanut smut, caused by Thecaphora frezzii Carranza & J.C. Lindq. (T. frezzii), is an emerging threat for the global peanut industry (BICON, 2017; Rago et al., 2017). First described in 1962, T. frezzii was observed on a collected sample of a wild peanut from Mato Grosso do Sul, Brazil, that was sent to INTA’s Manfredi Exp. Stn. in Córdoba Province, Argentina (Carranza & Lindquist 1962). In 1995, peanut smut was first detected on cultivated peanuts on three farms in north-central Córdoba (Marinelli et al., 2008). Since then, it has spread to all peanut-producing provinces in Argentina (Cazón et al., 2018). The disease’s destructive potential can be exemplified by pod incidence values as high as 70% (Bonessi et al. 2011, Chamberlin et al. 2022) and yield losses reaching 30% (Oddino et al. 2010). Disease management strategies using pesticides or biocontrol agents (Figueredo et al. 2017), liming (Bonessi et al. 2011), tillage (Cignetti et al. 2010; Cazón et al. 2014), and rotation (Marraro Acuña and Haro, 2011) have been attempted with modest results. Fungicides have shown moderate but highly variable control levels (Rago et al. 2017; Paredes et al. 2021). Smut resistant cultivars seem the best tool for disease management as they suffer less crop loss and limit the pathogen’s multiplication (Rago et al. 2017). Sources of smut resistance have been reported in wild peanuts (de Blas et al. 2019), synthetic amphidiploids (de Blas et al. 2021) and in A. hypogaea core collections (Chamberlin et al. 2022; Wann et al. 2020). Smut genetic resistance has shown high broad sense heritability (Bressano et al. 2019; de Blas et al. 2021). Recently, major and a minor quantitative trait loci (QTL) associated with moderate resistance have been identified in a recombinant inbred line (RIL) population developed from a cross between a susceptible A. hypogaea genotype and a resistant synthetic amphidiploid [A. correntina × A. cardenasii] × A. batizocoi (de Blas et al. 2021). Peanut reaction to T. frezzii is assessed by opening each pod by hand and inspecting it visually for incidence (i.e., the presence or absence of T. frezzii in pods), which is time and labor intensive. Screening for smut resistant genetic sources requires years of in-field trials along with pod phenotyping and is currently the only option for breeders because markers for resistance have not yet been developed.
Marker assisted selection (MAS) is now widely used in plant breeding to increase the efficiency of developing cultivars with highly desired traits. Early marker work identified several for disease resistance in peanut. Random amplified polymorphic DNA (RAPD) was used to identify root-knot nematode (RKN) resistance (Burow et al., 1996), restriction fragment length polymorphisms (RFLPs) were identified for resistance to Meloidogyne arenaria (Church et al., 2000), amplified fragment length polymorphisms (AFLPs) were found linked to resistance to the vector of groundnut rosette virus (Herselman et al., 2004), and single sequence repeats (SSRs) associated with resistance to Sclerotinia minor (Jagger) (Chenault, et al., 2009) along with those for tomato spotted wilt virus (TSWV) and leaf spot (LS) (Wang et al., 2013; Tseng et al., 2016; Zhao et al, 2018), rust (Khedikar et al., 2010; Mondal and Badigannavar, 2018), and bud necrosis disease (Jadhav et al., 2019) were identified.
Sequencing of the diploid progenitors (Bertioli et al, 2016), A duranensis and A. ipaensis, provided the reference genomes necessary to construct the high-density single nucleotide polymorphism (SNP) genotyping array Axiom_Arachis (Pandey et al., 2017) which led to the generation of SNP-based linkage maps enabling the identification SNPs associated with traits. SNPs are preferred over other marker types due to their wide distribution throughout the peanut genome (Liao and Lee, 2010). Whole genome resequencing (WGRS) of mapping populations has also been used to generate high-density SNP-based maps and identify QTL along with candidate genes for resistance to peanut diseases such as early LS (ELS), late LS (LLS), and spotted wilt (Agarwal et al., 2018). Such linkage maps have also been used to identify QTL for resistance in peanut to RKN (Leal-Bertioli et al., 2016), Aspergillus flavus (Khan et al., 2020), bacterial wilt (Luo et al, 2020a), stem rot (Luo et al. 2020b), Sclerotinia blight (Liang et al., 2021), and bud necrosis disease (Jasani et al., 2021). The recent release of the tetraploid (cultivated) peanut genome sequence (Bertioli et al., 2019) has further clarified questions regarding A. hypogaea evolution and the generation of a high-density SNP array for tetraploid peanut (Axiom_Arachis2) (Clevenger et al., 2017, Clevenger et al., 2018) provide further foundation for rapid marker-trait association. Using the Axiom_Arachis SNP array, potential SNPs and QTL associated with peanut smut resistance were identified by association mapping using a limited number of genotypes (Massa et al., 2021), but no confirmed QTL or candidate genes have been reported to date. Therefore, the objectives of this study were to perform whole genome sequencing (WGS) on a population developed for smut resistance mapping and subsequently fine map discovered QTL associated with smut resistance.
Materials and Methods
Plant Materials and Population Development.
The mapping population consisting of 200 lines was developed by crossing Mf10_2870, smut susceptible (J. Baldessari, unpublished data), and Ascasubi Hispano, smut resistant (Ibañez et al., 2018). An additional population used for validation purposes was also developed by crossing Granoleico, smut susceptible (Soave, 2002; Oddino et al., 2013) and Ascasubi Hispano. To rapidly identify QTL associated with smut resistance, a non-traditional approach was used where families within the population were phenotyped and genotyped at familial stages prior to the F7 generation rather than developing a uniform recombinant inbred line (RIL) population. Briefly, three individuals from each F2:3 family (for the mapping population) and from each F3:4 family (for the validation population) were phenotyped under heavily smut infested field conditions. This was done during the 2019-2020 season for the mapping population and during the 2020-2021 season for the validation population, in General Deheza, Córdoba Province, Argentina under rainfed conditions.
Experimental Design and Field Testing.
Smut resistance tests were conducted as described in Chamberlin et al., 2022, in fields adjacent to a farm (-32.759, -63.770) in the town of General Deheza (Córdoba Province, Argentina). Soil at the site was a General Deheza coarse-silty sandy loam type (0.3% slope, coarse-silty, mixed, thermic, Entic Haplustoll). The tests were planted one km downwind from a peanut processing plant where peanut smut is prevalent. The production field site was on a rotation schedule of corn-corn-soybean-peanut, where the preceding crop of the test site was always soybean.
Two control entries, one resistant (Ascasubi Hispano) and one susceptible (Colorado Irradiado INTA) were planted in both seasons. They were replicated twelve times each. Each family was represented by three individuals, each one constituting a plot. Plots were arranged in a square grid and randomized within it. Controls were placed, alternately, along a diagonal of the grid. Individual plots were spaced one m apart among rows and between plants.
The tests were planted on 11/7/19 and 12/23/20, using an augmented grid design with three replications, using single plants as experimental units. Plants were respectively dug at 147 and 140 days after planting (DAP), and all pods from each plant were harvested by hand and placed in mesh bags to air dry for two months. Yearly soil spore count was done as described by Chamberlin et al. (2022) to determine disease pressure. The test was conducted following the extension guidelines of the Instituto Nacional de Tecnología Agropecuaria (INTA).
Harvest and Evaluation of Smut Resistance.
Each pod was opened by hand and rated for disease incidence, i.e., presence or absence of T. frezzii sori or spore masses on the kernels. Disease incidence (DI) was calculated as DI = Infected pods / Total pods.
QTL mapping and validation.
DNA was taken from 572 individuals (approximately 3 individuals from each of the 200 lines) of the F2:3 family (Mf10_2870 and Ascasubi Hispano. DNA was also taken from 96 individual plants of F2:3 family of Ascasubi x Granoleico selected for phenotype. DNA extraction was performed from 20 mg of dried young leaves in silica gel. The samples were ground for two 10-sec cycles in bead mill at 15.000 rpm (Super FastPrep-2 Bead Beating System, MP BiomedicalsLLC, Irvine, CA, USA). A modified CTAB method was used with a sorbitol cleaning wash before the lysis step (Inglis et al. 2018). DNA quality was determined using 0.8% agarose gel and the DNA quantity was estimated with DS-11+ Nano UV-Vis Spectrophotometer (DeNovix Inc., DE, USA).
Parental genomic DNA extraction.
For whole-genome sequencing, seeds of Ascasubi INTA Cv, Granoleico Cv and Mf10_2870L were germinated in an incubation oven, under total darkness at constant temperature of 28 °C for 15 days. Fresh young etiolated leaves were collected for each genotype, immediately frozen with liquid nitrogen, and preserved at -80 C (Revco, Model ULT1386-5V41). The nucleii were extracted following the Nucleii Isolation – LN2 Plant Tissue Protocol (Ciculomics Inc./ PacBio) optimized from Workman et al. (2018). In the step 5 of protocol, filtration of the lysate with steriflip 20 µm pore was replaced with two lysate filtrations with a cell strainer of 100 µm pore and 40 µm pore. High-molecular weight (HMW) DNA was extracted from nucleii following the specifications of Nanobind Plant Nuclei Big DNA Kit (Circulomics Inc./ PacBio). DNA quality was determined using 0.8% agarose gel and the DNA quantity was estimated by Qubit V1 (Invitrogen ThermoFisher Scientific Inc.) using Qubit® dsDNA BR Assay Kits.
Genotyping: Affymetrix Axiom SNP Array.
Seeds from the peanut mini core accessions were sowed in 4.7-m pots with a mix of 50% Promix (Premier Tech Horticulture, Quaker, PA) and 50% steam-sterilized sandy soil from the Coastal Plain Experiment Station in Tifton, GA. Young leaf tissue was collected from 1-month-old plants for DNA extraction using DNeasy Plant mini kit ( www.qiagen.com). Quantification of DNA was performed with Quant-iT dsDNA assay kit ( www.thermofisher.com). DNA were submitted for genotyping with the Arachis version SNP array consisting of 47K SNPs features ( www.thermofisher.com). SNP data were curated from the Axiom analysis suite ( www.thermofisher.com). SNP markers were classified into six categories, i.e., PolyHighResolution, NoMinorHom, MonoHighResolution, CallRateBelowThreshold, OfftargetVariant, and Other according to the SNP QC matrix of the software (Clevenger et al., 2017). All data analysis in this study was performed with markers in the PolyHighResolution category due to its high quality in signal separation.
Genotyping: PacBio Sequencing
High molecular weight DNA was received from A. hypogea Granoleico and A. hypogea Ascasubi. DNA was sheared using the Diagenode Megaruptor 3 targeting 20kb fragments. Sheared DNA was prepared for PacBio sequencing using the SMRTbell Express Template Prep Kit 2.0. The library was size selected with AMpure PB beads to remove fragments less than 3kb. Sequencing was performed on a Sequel IIe System (Pacific Biosciences, Menlo Park, CA) using Binding Kit 2.2, Sequel II Sequencing Kit 2.0, and SMRTCell 8M. To target HiFi reads, the library was sequenced using a 30-hour movie time using Instrument Control Software Version 10. Raw subreads were converted to HiFi data by processing with CCS to call a single high quality consensus sequence for each molecule, using a 99.5% consensus accuracy cutoff. A. hypogea Granoleico was sequenced on 2 SMRTcells yielding 73.11Gb of HiFi data, and A. hypogea Ascasubi was sequenced on 3 SMRTcells yielding 61.37Gb of HiFi data.
Genotyping: Khufu whole-genome sequencing and analysis
DNA from populations were prepped into sequencing libraries using iGenomX Riptide library prep. For the F2:3 population, Six Riptide libraries, representing 576 samples were sequenced on 2 lanes of NovaSeq S4 chemistry (800 Gb raw base pairs per lane) to yield an average of 0.99X genome coverage per individual. The raw reads were demultiplexed and processed using Khufu (hudsonalpha.org/khufudata). To identify QTL for resistance, bulks were identified using one year of field data. Analysis of the bulks were done in silico, by bulking individual sequenced samples and calculating allele frequency differences between the putative resistant and susceptible bulks. The F3:4 samples were processed and analyzed the same way, except were sequenced to an average of 1.14X genome coverage. Haplotype analysis was done using HawkHAP, which is a component of Khufu.
Results and Discussion
Generation of reference genomes
Long read sequencing and assembly led to the generation of highly contiguous genome assemblies for smut resistant parent Ascasubi, and smut susceptible parent Granoleico (Table 1).
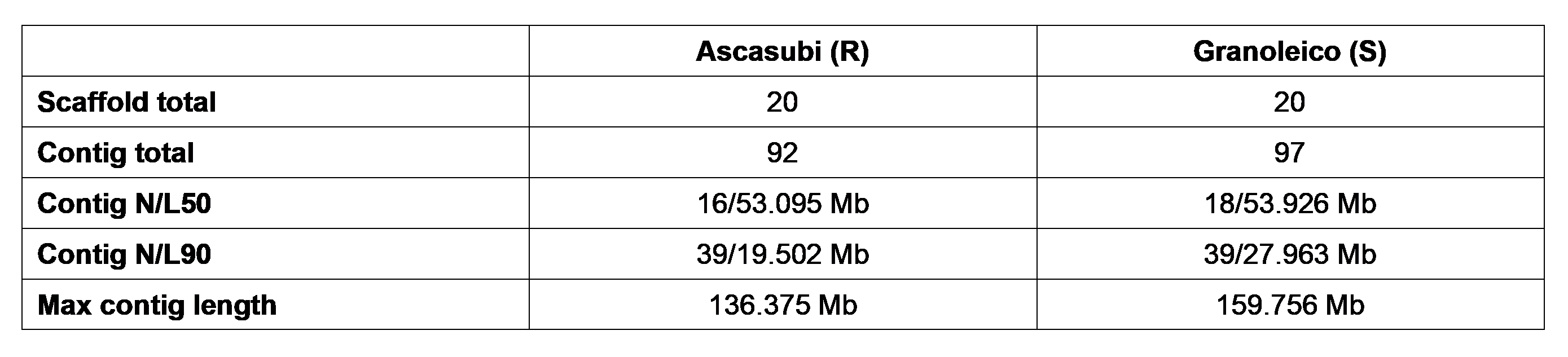
Assembly stats for the Ascasubi and Granoleico genome assemblies. Contigs were scaffolded into 20 pseudomolecules comprising the 10 A chromosomes and 10 B chromosomes. N/L50 is the length of the contig after half of the assembly has been accounted for when contigs are sorted from largest to smallest. Briefly, for Ascasubi half of the genome is assembled into 16 contigs that are longer than 53 Mb. N/L90 is calculated the same, but for 90% of the assembly. Briefly, for Ascasubi, 90% of the assembly is in 39 contigs which are longer than 19 Mb.
Mapping of smut resistance
A total of 576 individuals to construct bulks for mapping. They represented 200 F2:3 sub-families where each family was represented by 3 individuals. The smut pressure was low due to drought stress during pod development, but there were enough ‘susceptible’ individuals to map construct bulks (Figure 1). The frequency of infection was heavily skewed to the low end of the distribution, indicating that there was a considerable amount of false negative disease scores. To construct bulks, we employed a conservative approach whereby we selected susceptible individuals when all three sub-family members had infection and the average infection was greater than 5%. It was more difficult to select resistant bulks, because more than 400 individuals had 0% infection. For the ‘resistant’ bulk we selected every family member if they each had 0% infection and at least one individual was located in the field close to a susceptible check that exhibited infection. These criteria were chosen to reduce false negatives in the bulk as much as possible.
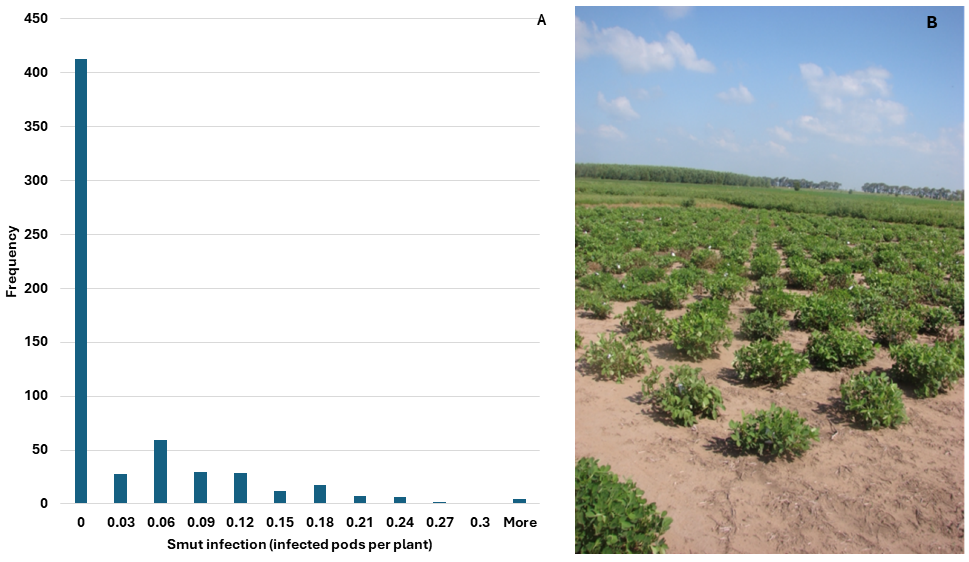
Figure 1. Smut infection as measured by infected pods/total pods in individual F2:3 progeny plants (A) tested at Manfredi in 2018-2019 field season (B).
We selected 45 individuals for the ‘resistant’ bulk and 49 individuals for the ‘susceptible’ bulk. The QTL-seq analysis was conducted by bulking bam alignment files in silico and assaying allele frequency differences genome-wide between the bulks (Figure 2A). Analysis of the bulks indicated one major QTL located on the proximal end of chromosome 12. There were no other peaks identified, indicating that a single, major locus accounted for smut resistance from Ascasubi. In the following year (2019-2020), we phenotyped a related population of F3:4 progeny with Ascasubi as a parent and Granoleico as the susceptible parent. We selected a set of 48 resistant individuals and the 48 most susceptible individuals. We sequenced those 96 individuals and again analyzed them with QTL-seq analysis. The major locus on chromosome 12 was again indicated as a single, major resistance locus controlling resistance (Figure 2B).
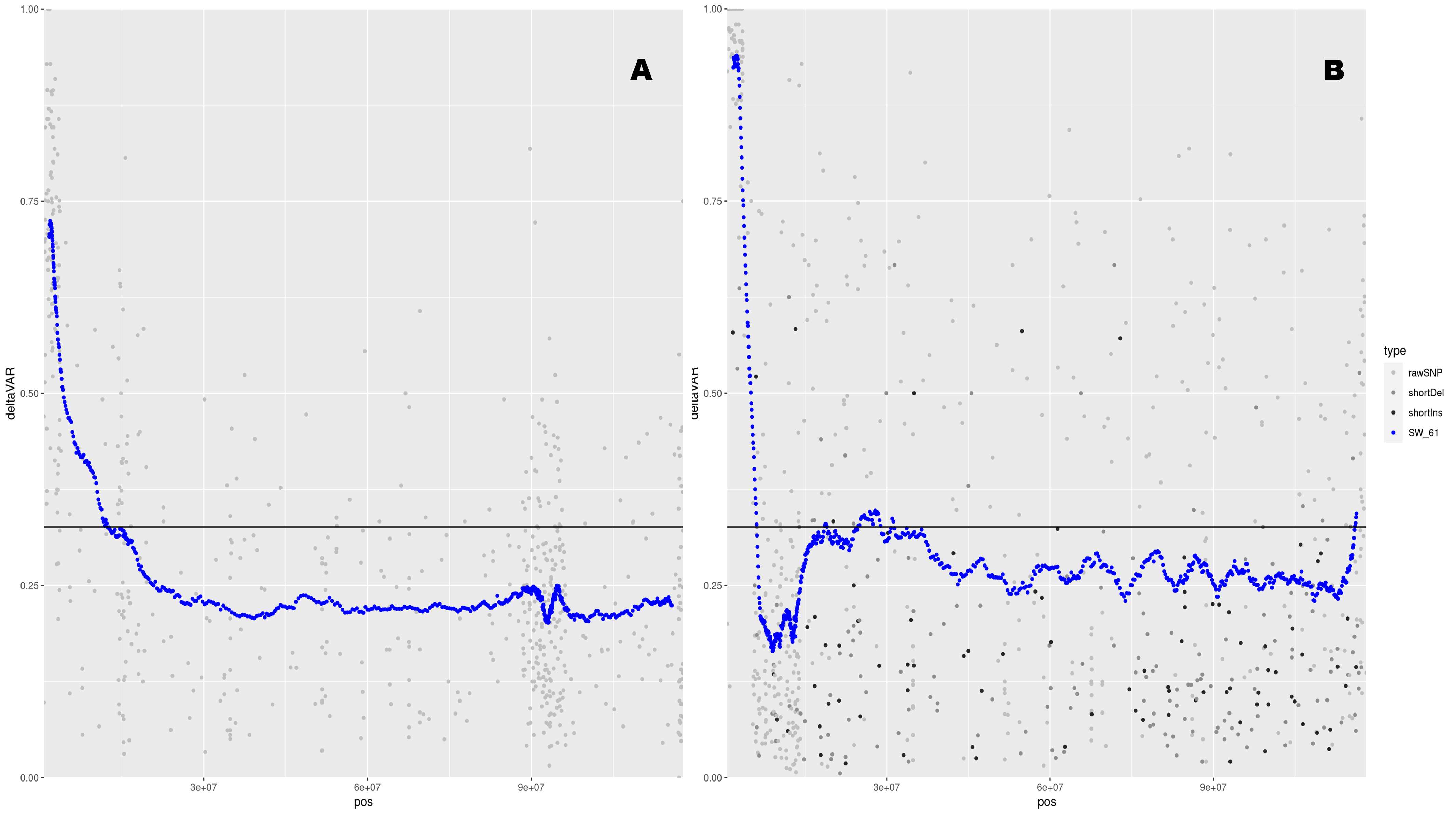
Figure 2. Mapping and validation of smut resistance. (A) A major QTL was identified on chromosome 12. Bulk analysis of 45 individuals with low infection and 49 individuals with infection >3% identifies a 2 Mb region conferring resistance. Y axis is the difference in allele frequency between the two bulks. A peak indicates a region where the allele frequencies significantly segregate between the bulks. The X axis is the physical location on the chromosome. (B) Validation of the QTL on chromosome 12 in a validation population of 48 resistant individuals and 48 susceptible individuals.
We have identified and validated a major locus controlling smut resistance in peanut. The qualitative inheritance of this trait, its high level of heritability, low environmental variance, and strong phenotyping methodologies, we were able to map and validate this resistance within early generations (F2:3 and F3:4), by phenotyping single plants rather than replicated plots. Phenotyping and genotyping early generation families within a developing population saves time and allows the potential for selection for backcross breeding. This method greatly decreased the time needed for discovery. The resistance is indicated on an approximal 2 Mb region that contains the largest cluster of R genes in the peanut genome (Bertioli et al., 2016). We have generated highly contiguous genome assemblies of the resistant parent, Ascasubi, and of the most widely grown cultivar in Argentina, Granoleico. Granoleico is very susceptible to smut, and the differences between those two genomes within the 2 Mb region will contain the functional variation that confers strong resistance to smut. This resistance and the markers associated with it will be transformational in breeding smut resistant cultivars for Argentina and the world.
Acknowledgements
Funding for this project was provided by USDA ARS CRIS Projects 3072-21220-008-00D, 6048-21000-029-000D, 6066-21310-005-000D, and 6046-21000-012-000D, the USDA ARS National Plant Disease Recovery System (NPDRS,) and MARS WRIGLEY, Inc. The authors declare no conflict of interest. The authors would like to thank Lisa Myers for technical assistance. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer.
Literature Cited
Agarwal G., Clevenger J., Pandey M.K., Wang H., Shasidhar Y., Chu Y., Fountain J.C., Choudhary D., Culbreath A.K., Liu X., Huang G., Wang X., Deshmukh R., Holbrook C.C., Bertioli D.J., Ozias-Akins P., Jackson S.A., Varshney R.K., and Guo B.. 2018. High-density genetic map using whole-genome re-sequencing for fine mapping and candidate gene discovery for disease resistance in peanut. Plant Biotechnol. J. DOI 10.1111/pbr.1274310.1111/pbi.12930.
Bertioli D.J., Cannon S.B., Froenicke L., Huang G., Farmer A.D., Cannon E.K.S., Liu X., Gao D., Clevenger J., Dash S., Ren L., Moretzsohn M.C., Shirasawa K., Huang W., Vidigal B., Abernathy B., Chu Y., Niederhuth C.E., , Araújo P., Kozik A.C.G., Kim A., Burow K.D., Varshney M.D., Wang R.K., Zhang X., Barkley X., Guimarães N., Isobe S., Guo B., Stalker H.T., Schmitz R.., Scheffler B .E., Leal-Bertioli S.C.M., Xun X., Jackson S.A., Michelmore R., and Ozais-Akins P.. 2016. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nature Genet. 48:438-446. DOI [: 10.1038/ng.3517].
Bertioli D.J., Jenkins J., Clevenger J., Dudchenko O., Gao D., Seijo G., Leal-Bertioli S.C.M., Ren L., Farmer A.D., Pandey M.K., Somoluk S.S., Abernathy B., Agarwal G. Ballén-Taborda C., Cameron C., Campbell J., Chavarro C., Chitikineni A., Chu Y., Dash S., El Baidouri M., Guo B., Huang W., Kim K.D., Korani W., Lanciano S., Lui C.G., Mirouze M., Mortensohn M.C., Pham M., Shin J.H., Shirasawa K., Sinharoy S., Sreedasyam A., Weeks N.T., Zhang X., Zheng Z., Sun Z., Froenicke L., Aiden E.L., Michelmore R., Varshney R.K., Holbrook C.C., Cannon E.K.S., Scheffler B.E., Grimwood J., Ozais-Akins P., Cannon S.B., Jackson S.A., and Schmutz J.. 2019. The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nature Genet. 51:877-884. DOI [: 10.1038/s41588-019-0405-z].
BICON. 2017. Australian Biosecurity Import Conditions. https://bicon.agriculture.gov.au/BiconWeb4.0/ViewElement/Element/Alert?elementPk=687363&casePk=682337.
Bonessi F., Rago A.M., Marinelli A.D., March G.J., Cazón L.I. García J., and Oddino C.M.. 2011. Efecto de la fertilización sobre la intensidad del carbon del maní. In:Jornada Nacíonal del Maní, General Cabrera, Córdoba, Argentina. pp. 63-64.
Bressano M., Massa A.N., Arias R.S., de Blas F., Oddino C., Faustinelli P.C., Soave S., Soave J.H., Pérez M.A., Sobolev V.S., Lamb M.C., Balzarini M., Buteler M.I., and Seijo J.G.. 2019. Introgression of peanut smut resistance from landraces to elite peanut cultivars (Arachis hypogaea L.). PLoS ONE 14: e0211920. DOI [: 10.1371/journal.pone.0211920].
Burow M.D., Simpson C.E., Paterson A.H., and Starr J.L.. 1996. Identification of peanut (Arachis hypogaea L.) RAPD markers diagnostic of root-knot nematode (Meloidogyne arenaria (Neal) Chitwood) resistance. Mol. Breed. 2:369-379. DOI [: 10.1007/BF00437915].
Carranza J. M., and Lindquist J.C.. 1962. Thecaphora frezii sp., parásita de Arachis sp. Boletín de la Sociedad Argentina de Botánica, X(1): 11–17.
Cazón L. I., Conforto C., Paredes J. A., Bisonard E. M., and Rago A.M.. 2014. Sensibilidad y especificidad de la técnica de PCR en la deteccíon de teliosporas de Thecaphora frezii en semillas de maní. In: 29° Jornada Nacional de Maní. General Cabrera, Córdoba, Argentina. pp. 40-42.
Cazón L.I., Paredes J.A., and Rago A.M.. 2018. The biology of Thecaphora frezii smut and its effects on Argentine peanut production. In J.N. Kimatu (ed.) Advances in plant pathology, IntechOpen Ltd., London, U.K. pp. 31–46. DOI 10.5772/intechopen.75837.
Chamberlin K.D., Baldessari J., Bennett R.S., Clevenger J.P., Holbrook C.C., Tallury S.P., Chu Y., Ozias-Akins P., Conde M.B., and Payton M.E.. 2022. Identification of germplasm resistant to peanut smut. Peanut Sci. 49: 1-16. DOI [: 10.3146/0095-3679-491-PS21-10].
Chenault K.D., Maas A.L., Damicone J.P., Payton M.E., and Melouk H.A.. 2009. Discovery and characterization of a molecular marker for Sclerotinia minor (Jagger) resistance in peanut. Euphytica 166:357-365.
Church G.T., Simpson C.E., Burow M.D., Paterson A.H. and Starr J.L.. 2000. Use of RFLP markers for identification of individuals homozygous for resistance to Meloidogyne arenaria in peanut. Nematology 2:575-580.
Cignetti M. I., Marraro Acuña F., and Mazzini P.H.. 2010. Influencia de la labranza sobre la intensidad del carbón del maní. Pages 12-14 in: 25 Jornada Nacional del Maní. General Cabrera, Córdoba, Argentina.
Clevenger J., Chu Y., Chavarro C., Agarwal G., Bertioli D.J., Leal-Bertioli S.C.M., Pandey M.K., Vaughn J., Abernathy B., Barkley N.A., Hovav R., Burow M., Nayak S.N., Chitikineni A., Isleib T.G., Holbrook C.C., Jackson S.A., Varshney R.K., and Ozias-Akins P.. 2017. Genome-wide SNP genotyping resolves signatures of selection and tetrasomic recombination in peanut. Mol. Plant 10(2):309–322 DOI 10.1016/j.molp.2016.11.015.
Clevenger J.P., Korani W., Ozias-Akins P., and Jackson S.. 2018. Haplotype-based genotyping in polyploids. Front in Plant Sci. 9:564 DOI 10.3389/fpls201800564.
Figueredo M.S., Tonelli M.L., Ibáñez F., Morla F., Cerioni G., Tordable M. C., and Fabra A.. 2017. Induced systemic resistance and symbiotic performance of peanut plants challenged with fungal pathogens and co-innoculated with the biocontrol agent Bacillus sp. CHEP5 and Bradyrhizobium sp. SEMIA6144. Microbiol. Res. 197: 65-73. DOI [: 10.1016/j.micres.2017.01.002].
Herselman L., Thwaites R., Kimmins F.M., Courtois B., van der Merwe P.J.A., and Seal S.E.. 2004. Identification and mapping of AFLP markers linked to peanut (Arachis hypogaea L.) resistance to the aphid vector of groundnut rosette disease. Theor. Appl. Genet. 109:1426-1433.
Ibañez M.A., Minudri F.H., Kearney M.I., Rago A.M., Paredes J.A., Mojica C., and Peiretti E.G.. 2018. Análisis multiambiental del comportamiento de genotipos de maní frente a carbón. Proc. "XXXIII Jornada Nacional de Maní", pp. 12. General Cabrera, Córdoba, Argentina. http://ciacabrera.com.ar/jornada_del_mani/33_jornada_del_mani.html.
Inglis P.W., Pappas Md.C.R., Resende L.V., and Grattapaglia D.. 2018. Fast and inexpensive protocols for consistent extraction of high-quality DNA and RNA from challenging plant and fungal samples for high-throughput SNP genotyping and sequencing applications. PLoS ONE 13(10): e0206085.DOI 10.1371/journal.pone.0206085.
Jadhav Y., Manohar S.S., Sunkad G., Kannalli V.P., Pandey M.K., Variath M.T., Yaduru S., Kona P., Varshney R.K. and Pasupuleti J.. 2019. Genomic regions associated with resistance to peanut bud necrosis disease (PBND) in a recombinant inbred line (RIL) population. Plant Breed. 138:748-760. DOI [: 10.1111/pbr.12743].
Jasani M.D., Kamdar J.H., Bera S., Sunkad G., and Bera S.K.. 2021. Novel and stable major QTLs conferring resistance to peanut bud necrosis disease and identification of resistant high yielding peanut breeding lines. Euphytica 217:105. DOI [: 10.1007/s10681-021-02835-7].
Khan S.A., Chen H., Deng Y., Chen Y., Zhang C., Cai T., Ali N., Mamadou G., Xie D., Guo B., Varshney R.K., and Zhuang W.. 2020. High-density SNP map facilitates fine mapping of QTLs and candidate genes discovery for Aspergillus flavus resistance in peanut (Arachis hypogaea). Theor. Appl. Genet. 133:2239-2257. DOI [: 10.1007/s00122-020-03594-0].
Khedikar Y.P., Gowda M.V., Sarvamangala C., Patgar K.V., Upadhyaya H.D., and Varshney R.K.. 2010. A QTL study on late leaf spot and rust revealed one major QTL for molecular breeding for rust resistance in groundnut (Arachis hypogaea L.). Theor. Appl. Genet. 121:971-84. DOI [: 10.1007/s00122-010-1366-x].
Leal-Bertioli S.C.M., Moretzsohn M.C., Roberts P.A., Ballén-Taborda C., Borba T.C., Valdisser P.A., Vianello R.P., Araújo A.C.G., Guimarães P.M., and Bertioli D.J.. 2016. Genetic mapping of resistance to Meloidogyne arenaria in Arachis stenosperma: a new source of nematode resistance for peanut. G3: Genes, Genomes, Genet. 6:377-390. DOI: 10.1534/g3.115.023044.
Liang Y., Cason J.M., Baring M.R., and Septiningsih E.M.. 2021. Identification of QTLs associated with Sclerotina blight resistance in peanut (Arachis hypogaea L.). Genet. Resour. Crop Evol. 68:629-637. DOI [: 10.1007/s10722-020-01012-4].
Liao P.Y. and Lee K.H.. 2010. From SNPs to functional polymorphism: the insight into biotechnology applications. Biochem. Eng. J. 49:149–158.
Luo H., Pandey M.K., Zhi Y., Zhang H., Xu S., Guo J., Wu B., Chen H., Ren X., Zhou X., Chen Y., Chen W., Huang L., Liu N., Sudini H.K., Varshey R.K., Lei Y., Liao B., and Jiang H.. 2020a. Discovery of two novel and adjacent QTLs on chromosome B02 controlling resistance against bacterial wilt in peanut variety Zhonghua 6. Theor. Appl. Genet. 133:1133-1148. DOI [: 10.1007/s00122-020-03537-9].
Luo Z., Cui R., Chavarro C., Tseng Y., Zhou H., Peng Z., Chu Y., Yang Z., Lopez Y., Tillman B., Dufault N., Brenneman T., Isleib T.G., Holbrook C., Ozias-Akins P., and Wang J.. 2020b. Mapping quantitative trait loci (QTLs) and estimating the epistasis controlling stem rot resistance in cultivated peanut (Arachis hypogaea). Theor. Appl. Genet. 133:1201-1212. DOI [: 10.1007/s00122-020-03542-y].
Marinelli A., March G. J., and Oddino C.. 2008. Aspectos biológicos y epidemiológicos del carbón del maní (Arachis hypogaea L.) causado por Thecaphora frezii Carranza & Lindquist. AgriScientia, 25: 1–5. DOI [: 10.31047/1668.298x.v25.n1.2735].
Marraro Acuña F. and Haro R.J.. 2011. Carbón del maní (Thecaphora frezii): su incidencia en rotaciones de cultivo. In: XXVI Jornada Nacional del Maní, Córdoba, Argentina, pp. 28-30
Massa A.N., Bressano M., Soave J.H., Buteler M.I., Seijo G., Sobolev V.S., Orner V.A., Oddino C., Soave S.J., Faustinelli P.C., de Blas F.J., Lamb M.C., and Arias R.S.. 2021. Genotyping tools and resources to assess peanut germplasm: smut-resistant landraces as a case study. Peer J. DOI 10.7717/peerj./10581.
Mondal S. and Badigannavar A.M.. 2018. Mapping of a dominant rust resistance gene revealed two R genes around the major Rust_QTL in cultivated peanut (Arachis hypogaea L.). Theor. Appl. Genet. 131:1671-1681. DOI [: 10.1007/s00122-018-3106-6].
Oddino C., Marinelli A., March G., García J., Tarditi L., D’Eramo L., and Ferrari S.. 2010. Relación entre el potencial inóculo de Thecaphora frezii. La intensidad de carbón del maní y el rendimiento del cultivo. 25 Jornada de Maní. General Cabrera, Córdoba. CIA-INTA. pp. 24–26.
Oddino C., Soave J., Soave S., Moresi A., Bianco C., Buteler M., Faustinelli P., and Torre D.. 2013. Avances genéticos en la tolerancia a carbón del maní causado por Thecaphora frezii. In: Proc. "XXVIII Jornada Nacional de Maní", pp. 31-32. General Cabrera, Córdoba, Argentina. http://ciacabrera.com.ar/jornada_del_mani/28_jornada_del_mani.html
Pandey M.K., Agarwal G., Kale S.M., Clevenger J., Nayak S.N., Sriswathi M., Chitikineni A., Chavarro C., Chen X., Upadhyaya H.D., Vishwakarma M.K., Leal-Bertioli S., Liang X., Bertioli D.J., Guo B., Jackson S.A. Ozais-Akins P., and Varshney R.K.. 2017. Development and evaluation of a high-density genotyping ‘Axiom_Arachis’ array with 58 K SNPs for accelerating genetics and breeding in groundnut. Sci. Rep. 7: 40577. DOI 10.1038/srep40577.
Paredes J.A., Cazón L.I., Oddino C., Monguillot J.H., Rago A.M. and Edwards Molina J.P.. 2021. Efficacy of fungicides against peanut smut in Argentina. Crop Prot. 140:105403. DOI [: 10.1016/j.cropro.2020.105403].
Rago A.M., Cazón L.I., Paredes J.A., Molina J.P.E., Conforto E.C., Bisonard E.M., and Oddino C.. 2017. Peanut smut: From an emerging disease to an actual threat to Argentine peanut production. Plant Dis. 100: 400-408, DOI [: 10.1094/PDIS-09-16-1248-FE].
Soave J.H. 2002. Granoleico, nuevo cultivar de maní (Arachis hypogaea L.) tipo runner con alta relacion oleico-linoleico. In: Proc. "XVII Jornada Nacional de Maní", pp. 40. General Cabrera, Córdoba, Argentina. http://ciacabrera.com.ar/jornada_del_mani/17_jornada_del_mani.html
Tseng Y., Tillman B.L., Peng Z., and Wang J.. 2016. Identification of major QTLs underlying tomato spotted wild virus resistance in peanut cultivar Florida-EPTM ‘113’. BMC Genet. 17:1. DOI [: 10.1186/s12863-016-0435-9].
Wang H., Pandey M.K., Qiao L., Qin L., Qin H., Culbreath A.K., He G., Varshney R.K., Scully B.T., and Guo B.. 2013. Genetic mapping and qualitative trait loci analysis for disease resistance using F2 and F5 generation-based genetic maps derived from ‘Tifrunner’ x ‘GT-C20’ in peanut. The Plant Gen. 6:1-10. DOI [: 10.3835/plantgenome2013.05.0018].
Wann D.Q., Falco A., Cavigliasso M., and Cassano C.. 2020. Phenotypic variation of peanut smut (Thecaphora frezii) incidence and severity in the U.S. peanut mini-core collection. Peanut Sci. 47: 46-53. DOI [: 10.3146/PS20-4.1].
Workman R., Timp W., Fedak R., Kilburn D., Hao S., and Liu K.. 2018. High Molecular Weight DNA Extraction from Recalcitrant Plant Species for Third Generation Sequencing. Protocol Exchange. DOI 10.1038/protex.2018.059.
Zhao Z. Tseng Y. Peng Z., Lopez Y., Chen C.Y., Tillman B.L., Dang P., and Wang J.. 2018. Refining a major QTL controlling spotted wild disease resistance in cultivated peanut (Arachis hypogaea L.) and evaluating its contribution to the resistance variations in peanut germplasm. BMC Genet. 19:17.
Notes
- USDA-ARS, Peanut and Small Grains Research Unit, Stillwater, Oklahoma [^]
- Instituto Nacional de Technologia Agropecuaria, Manfredi, Argentina [^]
- Aceitera, General Deheza, Argentina [^]
- USDA-ARS, Crop Genetics and Breeding Research Unit, Tifton, Georgia [^]
- National Environmentally Sound Production Agriculture Laboratory, Institute of Plant Breeding, Genetics, and Genomics, University of Georgia, Tifton, Georgia [^]
- USDA-ARS, Plant Genetic Resources Conservation Unit, Griffin, Georgia [^]
- HudsonAlpha Institute for Biotechnology, Huntsville, Alabama [^]
- USDA-ARS, Genomics and Bioinformatics Research Unit, Stoneville, Mississippi [^] Corresponding Author