

Introduction
The use of alien introgressions and substitution lines have been powerful tools for genetics and breeding. In wheat, alien introgressions from other grasses and rye have introduced virus resistance (Ali et al., 2016), fungal resistance (Kuraparthy et al., 2007; Lu et al., 2016), abiotic stress tolerance (Mohammed et al., 2013), increased yield (Zhang et al., 2015), and even haploid induction traits (Britt and Kuppu, 2016). Identification of these beneficial introgressions historically has been done using cytological methods such as in situ hybridization, but these methods have low resolution and are not high throughput (Lukaszewski et al., 2005). More advanced marker technologies, such as Simple Sequence Repeat (SSRs) and Restriction fragment length polymorphism (RFLP) markers increased the resolution and throughput of introgression identification (Eshed and Zamir, 1995; Fonceka et al., 2009). The power of ubiquitous and high throughput single nucleotide polymorphism (SNP) markers has led to efforts to provide SNP resources for alien introgression mapping in wheat (Tiwari et al., 2014). Wheat is only one example as alien introgression lines have been useful for many crops, including tomato (Eshed and Zamir, 1995), rice (Multani et al., 2003; Ray et al., 2016), cotton (Ulloa et al., 2016), and Brassica (Sharma et al., 2016).
Arachis hypogaea L. is an allotetraploid that has undergone an extreme genetic bottleneck and currently suffers from a narrow genetic base (Kochert et al., 1991). The diploid wild species of Arachis, however, harbor beneficial alleles for many important diseases (Stalker, 1984; Pande and Rao, 2001; Xue et al., 2004; Leal-Bertioli et al., 2015c). Efforts have been made to transfer these beneficial alleles into cultivated germplasm by creating induced allotetraploids or hexaploids (Stalker, 1984; Simpson, 1991; Simpson et al., 1993; Fonceka et al., 2012; Leal-Bertioli et al., 2015a; Stalker et al., 2013).
A highly successful introgression in A. hypogaea was a major resistance gene for root-knot nematode (Melodogyne arenaria) brought in from A. cardenasii by two separate groups (Simpson et al., 1993; Garcia et al., 1996). This source of resistance, released as germplasm in 1993 and 2002, has been utilized in cultivars to great success and is still durable in the field (Simpson et al., 1993; Stalker et al., 2002; Simpson et al., 2003; Holbrook et al., 2008). Selection of this resistance has been done using markers in many breeding programs and has successfully accelerated the breeding process (Chu et al., 2011). Attempts to fine map this resistance were stymied by recombination suppression and a lack of marker density (Nagy et al., 2010), and the marker used to select for resistance began to show broken linkage with the trait in breeding programs (Branch et al., 2014). Access to high throughput SNP markers allowed higher resolution mapping of the alien introgression which lead to development of a more tightly linked marker for use in marker-assisted selection programs (Chu et al. 2016).
Crowd-sourcing, which maximizes information gathered by collecting from many people (Greshake et al., 2014; Rallapalli et al., 2015; Li and Jackson, 2016), can be an efficient way to develop community resources. Although published data is deposited in public databases, it is in raw form, and must be processed by skilled personnel before its value can be realized. A crowd-sourcing tactic to increase SNP resources for introgression mapping will allow users to easily generate new data sources in a format that simplifies data sharing.
Here we report a new tool IntroMap that is an automated pipeline to map alien introgressions. The input for IntroMap is an alignment of your genotype of interest, reference genome, and diagnostic SNP set for the species/accession that has donated the introgression. IntroMap includes diagnostic SNP sets for five Arachis diploid species, but the pipeline includes a parameter to first develop diagnostic SNPs for the user's species/accession of interest. We use IntroMap to fine map alien introgressions in an elite A. hypogaea breeding line with superior disease resistance and demonstrate the performance at different sequence coverages. IntroMap can be a beneficial resource for all researchers using alien introgressions to incorporate beneficial alleles into elite germplasm in that it incorporates generated data into ready-to-use databases so that data sharing and accessibility efficiency increases for all researchers.
Materials and Methods
Sequence generation and processing
DNA from the diploid species A. cardenasii Krapov. & W.C. Gregory (GKP10017, PI262141) and A. diogoi (10602, PI 276235) was extracted from seedling leaves using a Qiagen DNeasy Plant mini kit® and sheared using Covaris® to obtain 550 bp insert size. Sequencing libraries were constructed using the TruSeq Nano DNA Library Prep Kit (www.illumina.com). The libraries were quantified using an Agilent DNA 7500 kit® on the Agilent 2100 Bioanalyzer. A high-output run of the Illumina NextSeq was completed at the Georgia Genomics Facility (Athens, GA; dna.uga.edu) generating 338,770,378 2 × 76 paired-end reads of A. cardenasii, 320,978,080 2 × 76 paired-end reads of A. diogoi, and 357,337,210 2 × 76 paired-end reads of IAC 322 (a tetraploid interspecific introgression line). DNAs from A. stenosperma Krapov. & W.C. Greg. (V10309, PI 666100) and A. magna Krapov., W.C. Gregory and C.E. Simpson (KG30097, PI 468340) were extracted using the DNeasy Plant Mini Kit (Qiagen). Three μg of DNA were used to construct PCR-free DNA libraries using the Kapa-Hyper Prep kit (Illumina ® Platforms). Libraries were sequenced using Illumina NextSeq (300 Cycles) PE150 High Output Flow Cell. RNA sequencing data was generated from pooled tissues of A. batizocoi (K9484, PI 298639). Reads were checked for quality using FastQC (Andrews, 2010) and trimmed using custom scripts.
Cleaned sequence from A genome species A. stenosperma, A. cardenasii, and A. diogoi, as well as IAC322 (to map an A genome introgression), was mapped to the A. duranensis pseudomolecules (Bertioli et al., 2016; peanutbase.org) as the A genome reference sequence, using BWA mem with default parameters (Li and Durbin, 2009). Cleaned sequence from B genome species A. magna and B genome compatible species A. batizocoi was mapped to the A. ipaënsis pseudomolecules (peanutbase.org) as the B genome reference sequence.
Identification of diagnostic SNP sets
To contrast the diploid-derived SNPs with A. hypogaea, SNPs were called using SAMtools mpileup (Li et al., 2009) with whole genome shotgun sequence from a set of 20 A. hypogaea accessions (Clevenger et al., 2016) mapped to either A. duranensis for A genome species or A. ipaënsis for B genome species. Mapping to either A or B genome was done to facilitate contrasting SNPs from either A or B genome compatible diploid species. These accessions represent the four market types of peanut. Arachis hypogaea L. subsp. hypogaea includes Runner and Virginia varieties (var. hypogaea) and A. hypogaea L. subsp. fastigiata includes Spanish (var. vulgaris) and Valencia (var. fastigiata) types. Also included is an accession representing A. hypogaea L. subsp. hypogaea var. hirsuta. Using custom scripts, SNPs in each diploid species were selected where all A. hypogaea accessions were determined to be homozygous for the reference base regardless of homeologous mapping. An additional criteria was applied that each SNP was not found in any of the other diploid species with compatible genomes. For example, a SNP in A. diogoi was compared to all other A genome species and was only considered diagnostic (or, in phylogenetic terms, autapomorphic) if it was a unique base to A. diogoi only. These sets of SNPs were compiled as diagnostic for their species. The SNP sets are accession-specific, but accessions analyzed in this study were historically used as parents of synthetic amphidiploids and included to maximize the relevance of the diagnostic SNP sets.
Sequence sub-sampling
To investigate how sequence coverage affects diagnostic SNP detection within introgressions, the sequence of IAC 322 was subsampled using seqtk sample (https://github.com/lh3/seqtk) for 1X, 2X, 4X, and 8x coverage of the A. hypogaea genome. Subsampled sequence was then processed as above for each coverage.
IntroMap dependencies
IntroMap uses SAMtools mpileup to call all possible SNPs before processing and bcftools to convert the BCF file to VCF. IntroMap is compatible with the latest versions of SAMtools and bcftools. These versions are included in the IntroMap package but need to be installed before use. When running IntroMap, it is necessary to add the statements to the shell script, "export PATH=/path/to/samtools/:$PATH" and "export PATH=/path/to/bcftools/:$PATH". IntroMap additionally requires Biopython which is also included with the package if not already on a user's system.
Results
IntroMap
IntroMap is a perl pipeline that uses SAMtools and custom python scripts to identify and map alien introgressions from diploid donors in A. hypogaea genotypes derived from inter-specific populations (Figure 1). The user supplies an alignment of a genotype of interest (e.g., introgression line) to the relevant reference genome (A. duranensis or A. ipaënsis). Sets of diagnostic SNPs for the donor species of the introgression are embedded in the pipeline. The pipeline calls SNPs using SAMtools for the genotype of interest relative to the user-supplied reference sequence and finds those SNPs that are in common with the diagnostic SNP set. The pipeline then checks each SNP in the genotype of interest for the correct diagnostic base and outputs the chromosome, SNP physical position, reference base, alternative base, and 100 bp on each side of the SNP for marker development (Supplemental Files 1 and 2). The density of diagnostic SNPs within introgressions clearly contrast these regions from noise (Figure S1). The pipeline does not make a judgement for introgression boundaries, but instead only reports the positions of diagnostic bases found within the genotype of interest. Introgressions show strong signal even at low sequence coverage (Figure S1A-E).
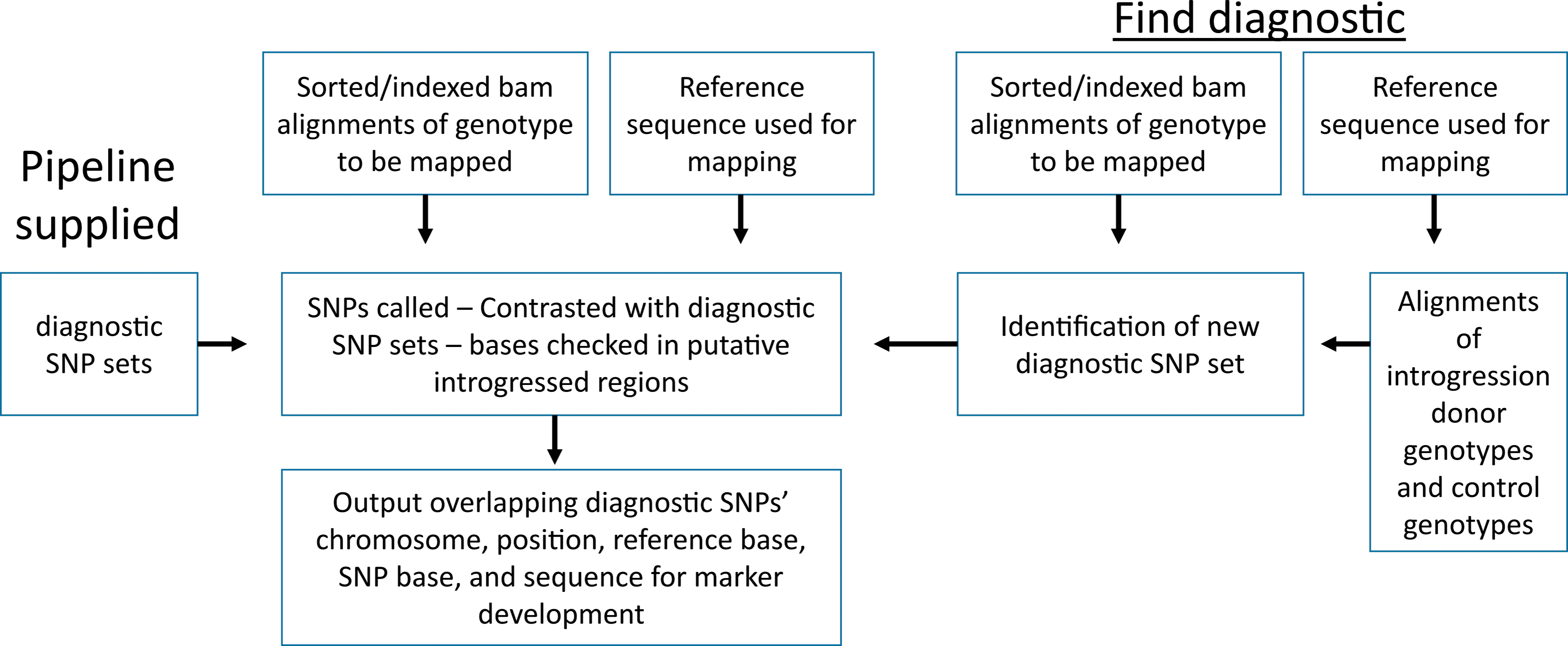
Pipeline overview. Input for the main pipeline is simply the sorted/indexed alignment file of the genotype of interest to a reference sequence and the reference sequence used. For Arachis hypogaea, the pipeline provides five different diploid species diagnostic SNP sets. Output is in tab delimited format of identified SNPs overlapping the diagnostic sets and 100 bp of sequence on each side for marker development. Using the parameter '-find_diagnostic', the user can include aligned sequence from the donor species and as many control genotypes as possible to contrast with the donor species. These newly identified SNPs are formatted for use in the pipeline and the main pipeline is automatically run to identify putative introgressions with the new SNP set.
Diagnostic SNP sets for Arachis
Five diploid Arachis species (Table 1) were used to construct diagnostic SNP sets for each species relative to the A. hypogaea progenitor species A. duranensis (A) and A. ipaënsis (B). These diploid species have been used as parents of current allotetraploids that have been used to introgress wild alleles into A. hypogaea and have been shown to exhibit beneficial traits related to resistance and tolerance to biotic and abiotic stresses (Stalker et al., 2002; Stalker et al., 2013; Leal-Bertioli et al., 2015a; Leal-Bertioli et al., 2015c). Genomic resequencing data were used for the A genome species A. cardenasii, A. diogoi, and A. stenosperma and the B genome species A. magna. RNA sequencing data was used for the B genome compatible species A. batizocoi. Table 1 shows the number of diagnostic SNPs that were identified for each species. All sets of diagnostic SNPs were contrasted with 21 A. hypogaea genotypes representing all four botanical types as well as the other diploid species represented.

Diagnostic SNPs identified for 5 Arachis species
User driven diagnostic SNP sets
A user can develop novel diagnostic SNP sets with the parameter '-find_diagnostic'. Use of this parameter triggers an additional pipeline that requires new parameters. These parameters are an alignment of the species/accession of interest along with at least one alignment of control genotypes from the species with the putative alien introgression that can be used to compare with the species/accession of interest. For example, to develop the diagnostic SNP sets from the five diploid wild Arachis species, 20 A. hypogaea genotypes were used as control. The more genotypes used for comparison, the higher confidence the user can have in the diagnostic SNPs identified. This pipeline identifies the diagnostic SNPs from the parameters included and generates the files in the format that IntroMap can use to map the alien introgressions of interest. The main pipeline then is automatically run with the newly generated diagnostic SNPs. The newly generated files with information on accession, species, and USDA PI number (if applicable) can then be sent to the IntroMap developers to be included in the pipeline for new users. As these resources increase, new users will not have to generate new data and the efficiency increases for all users.
Case Study: IAC 322
IAC 322 is a breeding line which was a progeny selection from the initial cross of Runner IAC 886, a popular cultivar in Brazil, and a germplasm line from ICRISAT (ICGV 86687: CS 16 - B2 - B2) that showed high levels of resistance to late leaf spot (Cercosporidium personatum) (I. Godoy, personal communication; ICRISAT, 1986). Because of the high level of resistance, which has only been observed in diploid species, it is suspected that IAC 322 inherited an alien diploid introgression. Because historically, the only known successful introgressions come from A. cardenasii (see above), this species was a candidate for use in this study. Genome sequence of line IAC 322 to 10x coverage of the A. hypogaea genome (2.8 Gb) was generated to test the efficacy of IntroMap for mapping potential introgressions from A. cardenasii. Three major introgressions were detected on chromosomes A02 and A03 (Table 2). The introgressions are between four and six million base pairs (Mb) in size. A diagnostic SNP identifying the introgressions occurs every 2.9 to 3.6 thousand base pairs (kb). The diagnostic SNPs are not the only SNPs identified in the region (Table 2), however there are no diagnostic SNPs shared with A. stenosperma. There are about five ambiguous SNPs for every diagnostic SNP in the introgressed regions. An ambiguous SNP is defined as an unshared SNP with the donor species that is also contrasting with A. hypogaea. The biological relevance of ambiguous SNPs is certainly important to discover, but can be disregarded for the purposes of this pipeline. The strength of the pipeline for accurate identification of introgressions derives from the extra step of checking each SNP with the diagnostic SNPs as well as checking the exact base change.
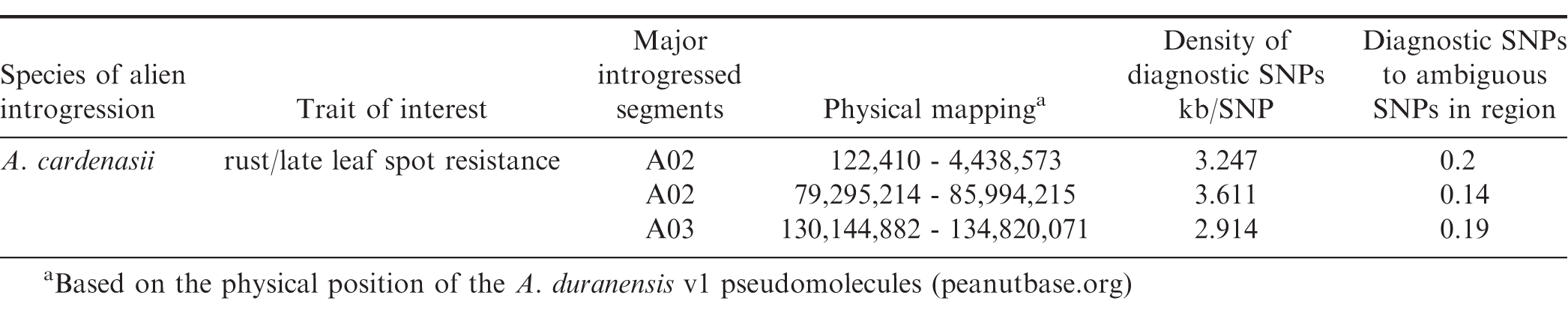
Three major A. cardenasii introgressions identified in breeding line IAC 322 with superior disease resistance.
As confirmation of the Intromap-identified introgressions of A. cardenasii, IAC 322 was genotyped using the Arachis 58K SNP array (Clevenger et al., 2016) along with A. cardenasii, A. duranensis, Runner 886, and a sister line (IAC 389). Markers that could contrast A. cardenasii from A. duranensis/A. hypogaea were selected. These markers confirmed the three major introgressions on chromosomes A02 and A03 (Table S1) as shared alleles between A. cardenasii and IAC 322 in these regions.
Sequence coverage
An important aspect to consider when carrying out experiments with Next Generation Sequencing data is how to balance the cost of the sequencing with sufficient sequence coverage for meaningful analysis. In a scenario where a researcher aims to map introgressions in an interspecific population, it may not be feasible to sequence each individual to 10X coverage. To investigate how IntroMap may perform with different sequence coverages, a subsampling experiment was performed (Figure 2). Using random subsampling at 1X, 2X, 4X, and 8X coverage, the IntroMap pipeline was run to test whether introgressions identified with 10X coverage were still detected at lower coverage. With 10X coverage set to 1, the percentage of SNPs identified with each coverage in total, in the A02 introgressions, and in the A03 introgressions was calculated. The amount captured was linear from 8-10X, with about 80%; of total SNPs, A02 SNPs, and A03 SNPs at 8X coverage. At lower coverages, however, the percentage of SNPs recovered was greater than expected. At 1X coverage, over 25%; of SNPs were recovered, and at 2X coverage almost 50%; of SNPs were recovered.
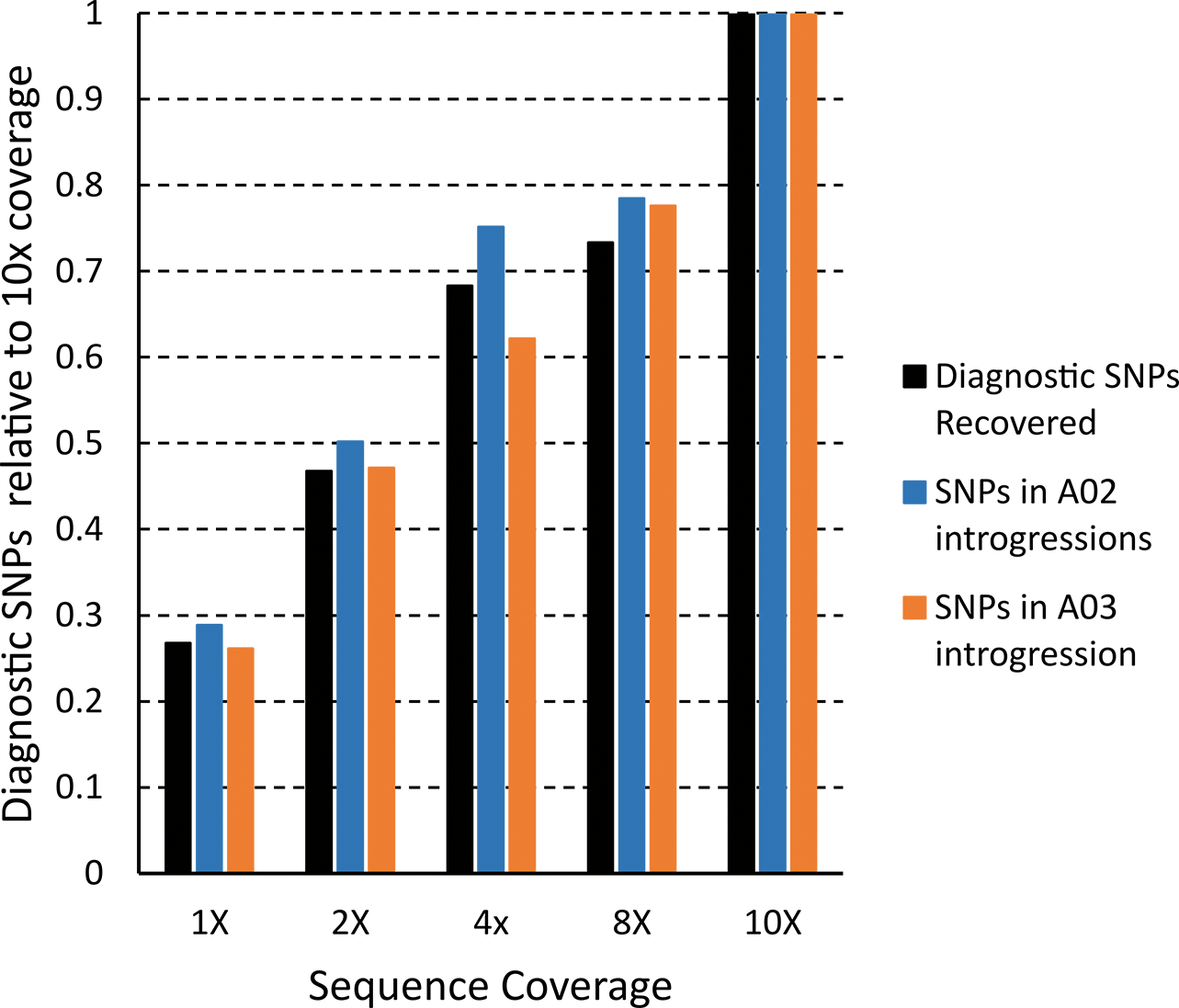
Effect of sequence coverage on introgression mapping. Total sequence of IAC 322 was sub sampled at indicated coverage levels and run through IntroMap. For each coverage the number of diagnostic SNPs found relative to 10X coverage is indicated for all SNPs (black), SNPs in the two major introgressions on A02 (blue), and the major introgression on A03 (orange).
IntroMap selects SNPs that are scored as homozygous alternate. At high sequence coverage, the probability that homeologous loci are not sequenced is lower than at lower coverages. As coverage decreases, certain loci that are ignored at higher coverage are called as introgression. This issue does not increase in a linear fashion as sequence coverage decreases. For example, as at 1X there are 2,001 of these ambiguous SNPs which is very similar to 2,075 at 2X coverage, and 1,708 at 8X coverage. Figure S1 (K-O) shows that the ambiguous noise does not prohibit the identification of true introgressions even at very low sequence coverage (1x).
Discussion
IntroMap is a one command, easy to use pipeline that can physically map alien introgressions. Although it was developed for Arachis species, IntroMap can be applied to any species where alien introgressions are used to incorporate beneficial alleles into elite germplasm. Five diploid Arachis species were used to identify diagnostic SNP sets that are included in the pipeline. One strength of this pipeline is its flexibility. All alien introgressions can be mapped if sequence of the donor accession/species of the introgression is available. There is an option to identify diagnostic SNP sets while using as many controls as may be available. Using this option, the pipeline will run a set of scripts that will identify the diagnostic SNPs, format them for use in the main pipeline, and run the main pipeline with that set of SNPs. All new sets of SNPs can then be shared to be incorporated with new releases of IntroMap. In this way it is truly a crowd-sourced tool that can grow and expand to be useful for many species and introgression lines.
Use of IntroMap in Arachis spp
The introgression of beneficial wild alleles into cultivated germplasm has resulted in the incorporation of beneficial traits for A. hypogaea that were not otherwise available (Stalker, 1984; Simpson et al., 1993; Simpson and Starr, 2001; Fonceka et al., 2012; Leal-Bertioli et al., 2015c; Favero et al., 2015). Simpson and Starr (2001) introgressed qualitative root-knot nematode (Melodogyne arenaria) resistance into A. hypogaea and released COAN. This resistance has since been used in several cultivars of importance and remains the sole source of major nematode resistance in A. hypogaea (Simpson et al., 2003; Holbrook et al, 2008; Branch and Brenneman, 2015). Fine mapping of introgressed segments has application as the introgression controlling the beneficial trait of interest is highly amenable to marker-assisted breeding. Using RNA sequencing analysis, the introgression controlling nematode resistance was fine mapped and led to the development of a new tightly linked marker that is deployed in marker-assisted selection programs (Clevenger et al., 2017).
Coverage and ambiguity
In experiments utilizing Next Generation Sequencing, there is always the question of adequate sequence coverage. For optimal efficiency, the minimum sequence coverage required to discover an introgressed segment is preferred in order to conserve resources for utilization in other areas of research rather than generating sequence that does not add informative data. Subsampling at lower coverages revealed that IntroMap can successfully identify major introgressions. Even as low as 1X coverage, the pipeline identifies over 25%; of diagnostic SNPs found using 10X coverage. Increasing the coverage to 2X identifies 50%; of the diagnostic SNPs (Figure 2). The diagnostic SNP density on an A02 introgression was one SNP every 3.2 kb with 10X sequence coverage (Table 2). For 1X coverage the density only decreased to a diagnostic SNP per 6.4 kb, and at 2X coverage the density is a comparable SNP per 4.3 kb. This result shows that IntroMap can be used with high confidence even when sequencing at low coverages. For a decrease in sequence of fivefold, IntroMap still identified close to 50%; of the diagnostic SNPs in the major introgressions as well as maintained a density that only decreased by 1 kb per SNP. An even further reduction in sequence coverage only slightly decreased those numbers to 25%; and a density depression of 3 kb per SNP.
One caveat with this approach is ambiguous regions that show signal for introgressions. At the highest coverage used in this study, only the three major introgressions were detected. As sequence coverage decreased, signals from across the genome began to appear. Manual inspection of a subsample of these satellite signals revealed that at high coverages, they were scored as 'heterozygous' and so were ignored by the pipeline. As the coverage decreased, some of these loci were sequenced only with the alternative base and so were scored as homozygous alternate. These bases represent an anomaly in that they were designated as specific polymorphisms only occurring in a specific species/accession. For the Arachis diagnostic SNPs developed in this study, they were compared against 20 diverse A. hypogaea accessions that represent the major groups of subsp. hypogaea var. hypogaea, subsp. hypogaea var. hirsuta, subsp. fastigiata var. vulgaris, and subsp. fastigiata var. fastigiata. The SNPs identified from each species were additionally compared against all other A and B genome compatible species used in this study. It is therefore unlikely that these ambiguous signals represent false positive diagnostic SNPs. The consequences of polyploidization can cause massive changes to genome structure and has been observed in Arachis previously (Ozkan et al., 2001; Leal-Bertioli et al., 2015b). The biological significance of this phenomenon is unknown. In the short term, it is a simple exercise to contrast the major introgressions from these signals at low coverages. The major introgressions show high density of diagnostic SNPs even at lower coverage levels and are easily distinguished from the less dense ambiguous SNPs which are more dispersed across the genome (1.15 Mb per SNP at 1X coverage).
Future prospects
IntroMap is truly a crowd-sourcing tool. In this study a database of Arachis diploid diagnostic SNP sets has been developed for access with the pipeline. The -find_diagnostic option will allow users to construct their own diagnostic SNP sets for their species/accession of interest and crop of interest. Novel SNP sets then can be added into the IntroMap pipeline. The power of this approach is to enable access to these SNPs sets without end-user investment in deep sequencing and data analysis. Certainly within the Arachis research community, there is a gradient in the abilities of labs to generate and analyze Next Generation Sequence data effectively. When sequence data are generated and published, raw data typically are deposited in databases for free distribution, but require downstream processing before use. The crowd-sourcing impact of IntroMap would be to compile these resources in a ready-to-use format from within the pipeline. The resources are developed within the pipeline itself upon use, requiring no additional processing.
Summary and Conclusions
Here we report the development of an automated pipeline for mapping alien introgressions in elite germplasm backgrounds. The pipeline should be applicable to map large introgressions from any species of interest with low whole genome sequence coverage. The pipeline currently includes diagnostic SNP sets for five diploid Arachis species that harbor beneficial alleles for disease resistance and abiotic stress. Included in the pipeline is an optional path that will extract diagnostic SNPs from sequence data from any species/accession of interest and use those SNPs for introgression mapping. These new SNP sets can then be included with the pipeline as a crowd-sourcing development of community resources. We show the efficacy of the pipeline using an A. hypogaea line that exhibits disease resistance that can be attributed to wild diploid alleles. IntroMap was used to identify three large introgressions with high resolution, even with sequence coverage as low as 1X. This has positive implications for mapping introgressions in populations when sequencing cost is prohibitive.
The value of IntroMap is accessibility. Any data that are generated from new species and used within the tool can be shared in a shared format. Reanalysis of raw data becomes unnecessary for each researcher wishing to use publically deposited raw data. Any species-specific sequence resources can be added to the pipeline so that as resources grow the accessibility also grows. Otherwise each researcher would have to identify robust diagnostic SNPs then write new scripts to conduct analysis. IntroMap is distributed freely, available at http://intromap.sourceforge.net.
Acknowledgements
This project was funded in part by the Agriculture and Food Research Initiative competitive grant 2012-85117-19435 of the USDA National Institute of Food and Agriculture (POA); the Peanut Foundation (POA, DJB) and the National Science Foundation BREAD program (project MCB-1543922; POA, SCMLB).
Literature Cited
Ali, N., J.P. Heslop-Harrison, H. Ahmad, R.A. Graybosch, G.L. Hein,and T. Schwarzacher. 2016. Introgression of chromosome segments from multiple alien species in wheat breeding lines with wheat streak mosaic virus resistance. Heredity 117: 114- 123.
Andrews, S. 2010. FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
D., Bertioli, S.B. Cannon, L. Froenicke, G. Huang, A.D. Farmer, E.K. Cannon, X. Liu, D. Gao, J. Clevenger, S. Dash, L. Ren, M.C. Moretzsohn, K. Shirasawa, W. Huang, B. Vidigal, B. Abernathy, Y. Chu, C.E. Niederhuth, P. Umale, A.C. Araújo, A. Kozik, Kim, K. Do M.D. Burow, R.K. Varshney, X. Wang, X. Zhang, N. Barkley, P.M. Guimarães, S. Isobe, B. Guo, B. Liao, H.T. Stalker, R.J. Schmitz, B.E. Scheffler, S.C. Leal-Bertioli, X. Xun, S.A. Jackson, R. Michelmore,and (). P. Ozias-Akins.
Branch, W., T.B. Brenneman,and G. Hookstra. 2014. Field test results versus marker assisted selection for root-knot nematode resistance in peanut. Peanut Sci. 41: 85- 89.
Branch, W.and T.B. Brenneman. 2015. Registration of 'Georgia-14N' peanut. J. Plant Registr. 9: 159- 161.
Britt, A.and S. Kuppu. 2016. Cenh3: An emerging player in haploid induction technology. Front. Plant Sci. 7: 357.
Clevenger, J., Y. Chu, C. Chavarro, G. Agarwal, D.J. Bertioli, S.C.M. Leal-Bertioli, M.K. Pandey, J. Vaughn, B. Abernathy, N.A. Barkley, R. Hovav, M. Burow, S.N. Nayak, A. Chitikineni, T.G. Isleib, C.C. Holbrook, S.A. Jackson, R.K. Varshney,and P. Ozias-Akins. 2016. Genome-wide SNP genotyping resolves signatures of selection and tetrasomic recombination in peanut. Molecular Plant 10: 309- 322.
Clevenger, J., Y. Chu, L. Guimaraes, T. Maia, D.J. Bertioli, S. Leal-Bertioli, P. Timper, C.C. Holbrook,and P. Ozias-Akins. 2017. Gene expression profiling describes the genetic regulation of Meloidogyne arenaria resistance in Arachis hypogaea and reveals a candidate gene for resistance. Sci. Rep. 7: 1317.
Chu, Y., C.L. Wu, C.C. Holbrook,and P. Ozias-Akins. 2011. Marker-assisted selection to pyramid nematode resistance and the high oleic trait in peanut. The Plant Genome 4: 110.
Chu, Y., R. Gill, J. Clevenger, P. Timper, C.C. Holbrook,and P. Ozias-Akins. 2016. Identification of rare recombinants leads to tightly linked markers for nematode resistance in peanut. Peanut Sci. 43: 88- 93.
Eshed, Y.and D. Zamir. 1995. An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTL. Genetics 141: 1147- 116.
Fávero, A., R.F. Dos Santos, C.E. Simpson, J.F. Valls,and N.A. Vello. 2015. Successful crosses between fungal-resistant wild species of Arachis (section Arachis) and Arachis hypogaea. Genet. Mol. Biol. 38: 353- 365.
Foncéka, D., T. Hodo-Abalo, R. Rivallan, I. Faye, M.N. Sall, O. Ndoye, A.P. Fávero, D.J. Bertioli, J.-C. Glaszmann, B. Courtois, J.-F. Rami. 2009. Genetic mapping of wild introgressions into cultivated peanut: a way toward enlarging the genetic basis of a recent allotetraploid. BMC Plant Biol. 9: 103- 110.
Foncéka, D., H.A. Tossim, R. Rivallan, H. Vignes, I. Faye, O. Ndoye, M.C. Moretzsohn, D.J. Bertioli, J.C. Glaszmann, B. Courtois,and J.-F. Rami. 2012. Fostered and left behind alleles in peanut: interspecific QTL mapping reveals footprints of domestication and useful natural variation for breeding. BMC Plant Biol. 12: 26.
Garcia, G.M., H.T. Stalker, E. Shroeder,and G. Kochert. 1996. Identification of RAPD, SCAR, and RFLP markers tightly linked to nematode resistance genes introgressed from Arachis cardenasii into Arachis hypogaea. Genome 39: 836- 845.
Greshake, B., P.E. Bayer, H. Rausch, J. Reda. 2014. OpenSNP-a crowdsourced web resource for personal genomics. PLoS One 9: e89204.
Holbrook, C.C., P. Timper, A.K. Culbreath,and C.K. Kvien. 2008. Registration of 'Tifguard' peanut. J. Plant Registr. 2: 92- 94.
ICRISAT Annual Report. 1986. Retrieved from http://agropedialabs.iitk.ac.in/openaccess/sites/default/files/Annual_Report_1986.pdf. 03 May 2017.
Kochert, G., T. Halward, W.D. Branch,and C.E. Simpson. 1991. RFLP variability in peanut (Arachis hypogaea L.) cultivars and wild species. Theor. Appl. Genet. 81: 565- 570.
Kuraparthy, V., P. Chhuneja, H.S. Dhaliwal, S. Kaur, R.L. Bowden,and B.S. Gill. 2007. Characterization and mapping of cryptic alien introgression from Aegilops geniculata with new leaf rust and stripe rust resistance genes Lr57 and Yr40 in wheat. Theor. Appl. Genet. 114: 1379- 1389.
Leal-Bertioli, S., S.P. Santos, K.M. Dantas, P.W. Inglis, S. Nielen, A.C. Araujo, J.P. Silva, U. Cavalcante, P.M. Guimãres, A.C. Brasileiro, N. Carrasquilla-Garcia, R.V. Penmetsa, D. Cook, M.C. Moretzsohn,and D.J. Bertioli. 2015 a. Arachis batizocoi: a study of its relationship to cultivated peanut (A. hypogaea) and its potential for introgression of wild genes into the peanut crop using induced allotetraploids. Ann. Bot. 115: 237- 249.
Leal-Bertioli, S., K. Shirasawa, B. Abernathy, M. Moretzsohn, C. Chavarro, J. Clevenger, P. Ozias-Akins, S. Jackson,and D. Bertioli. 2015 b. Tetrasomic recombination is surprisingly frequent in allotetraploid Arachis. Genetics 199: 1093- 1105.
Leal-Bertioli, S., M.C. Moretzsohn, P.A. Roberts, C. Ballén-Taborda, T.C. Borba, P.A. Valdisser, R.P. Vianello, A.C. Araújo, P.M. Guimarães,and D.J. Bertioli. 2015 c. Genetic mapping of resistance to Meloidogyne arenaria in Arachis stenosperma: A new source of nematode resistance for peanut. G3 (Bethesda) 6: 377- 390.
Li, H.and R. Durbin. 2009. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25: 1754- 1760.
Li, H., B. Handsaker, A. Wysoker, T. Fennell, J. Ruan, N. Homer, G. Marth, G. Abecasis, R. Durbin,and 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078- 2079.
Li, Y.and S.A. Jackson. 2016. Crowdsourcing the nodulation gene network discovery environment. BMC Bioinformatics 17: 223.
Lu, Y., M. Yao, J. Zhang, L. Song, W. Liu, X. Yang, X. Li,and L. Li. 2016. Genetic analysis of a novel broad-spectrum powdery mildew resistance gene from the wheat-Agropyron cristatum introgression line Pubing 74. Planta 244: 713- 723.
Lukaszewski, A., B. Lapinski,and K. Rybka. 2005. Limitations of in situ hybridization with total genomic DNA in routine screening for alien introgressions in wheat. Cytogenet. Genome Res. 109: 373- 377.
Mohammed, Y., A.E. Eltaye,and H. Tsujimoto. 2013. Enhancement of aluminum tolerance in wheat by addition of chromosomes from the wild relative Leymus racemosus. Breeding Sci. 63: 407- 416.
Multani, D., G.S. Khush, B.G. de los Reyes,and D.S. Brar. 2003. Alien genes introgression and development of monosomic alien addition lines from Oryza latifolia Desv. to rice, Oryza sativa L. Theor. Appl. Genet. 107: 395- 405.
Nagy, E., Y. Chu, Y. Guo, S. Khanal, S. Tang, Y. Li, W.B. Dong, P. Timper, C. Taylor, P. Ozias-Akins, C.C. Holbrook, V. Beilinson, N.C. Nielsen, H.T. Stalker, S.J. Knapp. 2010. Recombination is suppressed in an alien introgression in peanut harboring Rma, a dominant root-knot nematode resistance gene. Mol. Breed. 26: 357- 370.
Ozkan, H., A.A. Levy,and M. Feldman. 2001. Allopolyploidy-induced rapid genome evolution in the wheat (Aegilops-Triticum) group. Plant Cell 13: 1735- 1747.
Pande, S.and J.N. Rao. 2001. Resistance of wild Arachis species to late leaf spot and rust in greenhouse trials. Plant Dis. 85: 851- 855.
Rallapalli, G., Fraxinus Players, D.G. Saunders, K. Yoshida, A. Edwards, C.A. Lugo, S. Collin, B. Clavijo, M. Corpas, D. Swarbreck, M. Clark, J.A. Downie, S. Kamoun, Team Cooper,and D. MacLean. 2015. Lessons from Fraxinus, a crowd-sourced citizen science game in genomics. Elife 29: e07460.
Ray, S., L.K. Bose, J. Ray, U. Ngangkham, J.L. Katara, S. Samantaray, L. Behera, M. Anumalla, O.N. Singh, M. Chen, R.A. Wing,and T. Mohapatra. 2016. Development and validation of cross-transferable and polymorphic DNA markers for detecting alien genome introgression in Oryza sativa from Oryza brachyantha. Mol. Genet. Genomics 4: 1783- 1794.
Sharma, B., P. Kalia, D.K. Yadava, D. Singh,and T.R. Sharma. 2016. Genetics and molecular mapping of black rot resistance locus Xca1bc on chromosome B-7 in Ethiopian mustard (Brassica carinata A. Braun). PLoS One 11: e0152290.
Simpson, C. 1991. Pathways for introgression of pest resistance into Arachis hypogaea L. Peanut Sci. 18: 22- 26.
Simpson, C., S.C. Nelson, J.L. Starr, K.E. Woodard,and O.D. Smith. 1993. Registration of TxAG-6 and TxAG-7 peanut germplasm lines. Crop Sci. 33: 1418.
Simpson, C.and J.L. Starr. 2001. Registration of 'Coan' peanut. Crop Sci. 41: 918.
Simpson, C., J.L. Starr, G.T. Church, M.D. Burow,and A.H. Paterson. 2003. Registration of 'NemaTAM' peanut. Crop Sci. 43: 1561.
Stalker, H. 1984. Utilizing Arachis cardenasii as a source of cercospora leafspot resistance for peanut improvement. Euphytica 33: 529- 538.
Stalker, H.T., M.K. Beute, B.B. Shew,and K.R. Barker. 2002. Registration of two root-knot nematode-resistant peanut germplasm lines. Crop Sci. 42: 314- 316.
Stalker, H., S.P. Tallury, P. Ozias-Akins, D. Bertioli,and S.C. Leal Bertioli. 2013. The value of diploid peanut relatives for breeding and genomics. Peanut Sci. 40: 70- 88.
Tiwari, V., S. Wang, S. Sehgal, J. Vrána, B. Friebe, M. Kubaláková, P. Chhuneja, J. Doležel, E. Akhunov, B. Kalia, J. Sabir,and B.S. Gill. 2014. SNP Discovery for mapping alien introgressions in wheat. BMC Genomics 15: 273.
Ulloa, M., C. Wang, S. Saha, R.B. Hutmacher, D.M. Stelly, J.N. Jenkins, J. Burke,and P.A. Roberts. 2016. Analysis of root-knot nematode and fusarium wilt disease resistance in cotton (Gossypium spp.) using chromosome substitution lines from two alien species. Genetica 144: 167- 179.
Xue, H T.G. Isleib, H.T. Stalker,and G. O'Brian. 2004. Evaluation of Arachis species and interspecific tetraploid lines for resistance to aflatoxin production by Aspergillus flavus. Peanut Sci. 31: 134- 141.
Zhang, J., J. Zhang, W. Liu, H. Han, Y. Lu, X. Yang, X. Li, L. Li. 2015. Introgression of Agropyron cristatum 6P chromosome segment into common wheat for enhanced thousand-grain weight and spike length. Theor. Appl. Genet. 128: 1827- 1837.
Notes
- Department of Horticulture and Institute of Plant Breeding, Genetics and Genomics, The University of Georgia, Tifton, GA 31793, USA [^]
- Center for Applied Genetic Technologies and Institute of Plant Breeding, Genetics and Genomics. The University of Georgia. Athens, GA 30602, USA [^]
- University of Brasília, Institute of Biological Sciences, Campus Darcy Ribeiro, 70910-900 Brasília, DF, Brazil [^]
- Embrapa Genetic Resources and Biotechnology, 70770-917 Brasília, DF, Brazil [^]
- Dept. of Crop and Soil Sciences, Box 7629, N.C. State Univ., Raleigh, NC. 28695-7629 [^] *Corresponding author's Email: pozias@uga.edu
Author Affiliations